
金刚烷,其中金刚烷(C\(_{10}\)H\(_{16}\))是最简单的代表,构成了一类有趣的碳基纳米材料,具有有趣的化学,机械和光电性质。虽然中性金刚石已经被广泛研究了几十年,但它们的阳离子对偶物是最近实验研究的一个主题,其动机是它们在天体化学中的潜在作用。在这里,我们进行了金刚烷阳离子(C\(_{10}\)H\(_{16}^+\))的计算研究,以补充最近的实验发现。具体地说,我们通过考虑吸收光电子能谱中阳离子的高电子激发态,扩展了振动分辨电子能谱的早期理论工作。我们还进行了绝热和非绝热(表面跳跃)分子动力学模拟,以研究C\(_{10}\)H\(_{16}^+\)的(快速)碎裂过程和电子弛豫。我们的模拟表明,在近红外紫外光子激发后,金刚烷阳离子经历了一个超快的内部转换到基态(双重态)(在10-100 fs的时间尺度上,取决于初始激发能量),随后可以快速破碎,主要是H损失。报道了超快氢解离的产率与激发能的关系。
在自然界和星际化学中存在不同类型的碳适应[1,2,3]。一类有趣的碳化合物是具有类金刚石、氢饱和分子结构的类金刚石[4]。这些分子在纳米技术[5,6]、生物化学[7]以及药物化学和制药工业[8,9,10,11]中具有潜在的各种应用。它们具有良好的化学和热稳定性,这是科学界研究它们的一个主要因素[4,12]。天然存在于天然气和原油中的小类金刚石含量很高[13]。纳米金刚石(由100个碳原子组成的大型金刚石)构成了碳质原始球粒陨石[2]。最近在星际介质(ISM)中检测到巴克敏斯特富勒烯(C)[14,15,16],有人推测在ISM中存在更多大尺寸的碳衍生物[17]。
由于天体物理学的联系和纳米技术的相关性,人们也做了相当多的实验和理论研究来详细研究金刚石类化合物的光谱特性。例如,中性金刚石的红外(IR)光谱[18,19]、光吸收光谱[20]和荧光光谱[21]已被记录。光电子能谱也有报道[22,23,24]。第一性原理计算已被应用于计算原始和改性金刚石的(振动分辨)光谱[25,26,27,28]以及获得光电子能谱[28,29,30]。此外,还模拟了金刚烷[31]和含金刚烷化合物[32,33,34]的非绝热动力学。
最小的类金刚石是金刚烷分子(Ada, CH)[35]。虽然Ada已经被仔细研究了几十年[35,36,37,38],但由于其在ISM中的潜在作用,其阳离子(Ada, CH)最近引起了相当大的兴趣[39]。Ada的光谱是由Crandall等人[39]和Kappe等人[40]分别利用电子光解离(EPD)光谱和氦标记电子光谱实验获得的。此外,最近,Crandall等人报道了利用EPD获得金刚石烷阳离子(Dia)的光谱[41]。计算上,利用时间依赖密度泛函理论(TD-DFT)[39,42,43,44]和运动方程耦合簇单双(EOM-CCSD)[40]研究了Ada的垂直电子跃迁。除了垂直激发外,我们小组还计算了Ada的低能量、振动分辨吸收谱[44]和Ada的光电子谱[30]。
此外,Crandall等人[39]的EPD研究提供了光诱导Ada碎裂的信息。特别是,发现H损失通道在低激发能(相对于阳离子基态的外观能为1.38 eV)下占主导地位。此外,Candian等人用真空紫外(VUV)光子激发Ada,用成像光电子光离子重合(iPEPICO)光谱研究了Ada的解离[45]。据报道,H损失通道的外观能量相对于中性基态为10.50 eV(或相对于阳离子基态为1.21 eV)[45]。此外,Ada的碎片化已经通过电子电离质谱(EIMS)进行了检测[3,46]。我们还注意到,除了碎片化,金刚烷的相关化合物金刚烷阳离子的笼型打开,最近进行了研究[47,48]。
在这个计算研究中,我们追求几个方向来研究Ada。首先,我们扩展了之前的光谱模型[30,44],在振动分解吸收光谱和光电子光谱中考虑了阳离子的高电子激发态(尽管使用了下面描述的简单垂直梯度近似)。其次,我们对Ada进行了分子动力学模拟,仅依赖于基重态,并提供一定的多余能量,以揭示快速破碎过程。第三,我们利用表面跳变(SH)方法对Ada的超快非绝热动力学(内部转换)进行了建模。
利用一系列半经验方法(AM1[49]、PM3[50]、PM7[51]、OM2[52]和OM3[53])和B3LYP [54,55]/ 6-31G *[56,57]水平的DFT对金刚烷中性(Ada)和阳离子(Ada)分别在基电子态(和)进行了优化。中性离子采用自旋受限(R)形式,而阳离子的几何结构采用自旋不受限(U)形式。此外,在半经验构型相互作用单双能级(CISD)水平上对阳离子几何形状进行了优化,用于表面跳跃模拟(见下文)。特别地,CISD计算的分子轨道是通过限制开壳(RO) OM3计算得到的,CISD计算是在9个已占据轨道和10个虚轨道的限制有效空间中进行的。我们称此方法为ROOM3/CISD()。对于所有优化的结构,执行正态模态分析以确认所发现的平稳点的最小性质(即没有虚频率)。
利用TD- ub3lyp / 6-31G *级的线性响应时间相关(TD) DFT以及ROOM3/CISD()方法计算了阳离子电子跃迁的垂直激发能和相应的振荡器强度。这些计算是在用UB3LYP/ 6-31G *和ROOM3/CISD()优化的阳离子的基态几何()下进行的。此外,在优化的中性(最小)几何形状下,在TD-DFT水平上计算垂直跃迁,以估计垂直光电子能谱的跃迁能量(通过将垂直电离势添加到垂直激发能中)。
此外,为了计算阳离子的振动分辨吸收谱和振动分辨光电子谱,我们试图优化阳离子的激发态,但我们无法找到激发态的最小值。因此,我们采用了垂直梯度(VG)近似[58],该近似通过移动初始状态(在阳离子吸收和光电子能谱的情况下)的谐波PES,基于初始状态最小几何形状的最终状态梯度,构建最终状态()势能面(PES)。对于转换,我们还使用了绝热Hessian (AH)方法,该方法需要初始和最终状态的正常模式,反过来,由于成功地优化了两个状态,我们能够获得正常模式。光电子能谱使用时无关(TI)形式(基于frank - condon (FC)因子的计算)计算,吸收光谱使用时相关(TD)形式(基于自相关函数的计算)计算,因为TI形式导致一些考虑的跃迁的低光谱级数[58]。在FC近似下计算了所有振动分辨谱。使用默认的高斯16参数。特别地,采用了半宽为135 cm的高斯展宽。
此外,我们对金刚烷阳离子进行了从头开始的分子动力学模拟,重点是可能的断裂。为此,我们考虑了三种场景,如图1所示,分别标记为(a)、(b)和(c)。
图1
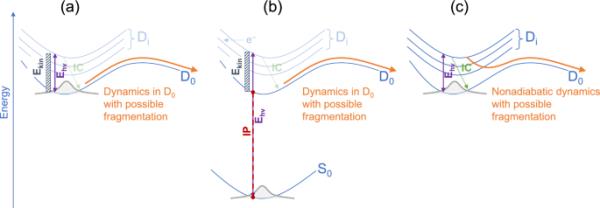
在这项工作中使用的建模方法的示意图。一种模拟阳离子在紫外光激发后电子光解离(EPD)的近似方法。光子能量被设为等于核动能。b模拟由VUV激发中性(从状态)引起的解离电离的近似方法。光子能量等于原子核动能加上电离势(IP)。c模拟紫外线激发后阳离子弛豫的精确方法。在a和b中,在状态下进行了绝热动力学模拟,而在c中进行了非绝热动力学模拟。在a和b中褪色的部分显示了在这些简化方法中没有明确说明的过程,例如内部转换(IC)和电子损失。详情见正文
情景(a)和(b)分别对应于EPD和VUV (EIMS)技术,两者都是在省略了明确处理阳离子激发态的近似下,因此没有明确描述内部转换(IC)过程。在(a)中,假设原本处于该状态的阳离子被紫外(或可见光、近红外)激光激发,光子能量转化为核动能,阳离子保持在基态()。在(b)中,假设原本处于该状态的中性被VUV光子或电子束激发,激发能中有一部分用于电离(电离势),其余部分转化为原子核动能,阳离子处于基态()。出射电子的动能被假定为零(如阈值光电子-光子符合(TPEPICO)光谱学)。然后在阳离子基态下进行Born-Oppenheimer分子动力学(BOMD)模拟。由于在使用不受限制方法的测试计算中发现了能量守恒问题,因此使用费米涂抹(在某种程度上对应于电子激发)。在费米涂抹法中,允许分子轨道占据是分数阶的,并使用费米-狄拉克分布计算,电子温度作为参数[59]。在我们的模拟中使用了默认值20,000 K(在MNDO程序[60]中)。我们注意到方法(a)和(b)类似于Grimme的QCEIMS(量子化学EIMS)[61]方法。在我们的工作中,UOM3方法被用于动力学模拟,因为它被发现产生的金刚烷电离势与实验很好地吻合(见下文)。利用速度Verlet算法以0.5 fs的时间步长传播10 ps的轨迹。分子的整体平移和旋转被消除,只关注内部动力学。轨迹以恒定的总能量传播。
在与EPD(类似于(a))对应的情形(c)中,明确考虑了阳离子激发态,并模拟了光激发后的非绝热动力学。因此,在方法(c)中直接考虑了IC。具体而言,这里使用Tully的表面跳变(SH)方法进行非绝热动力学模拟[62]。阳离子的电子结构用上文介绍的半经验ROOM3/CISD()方法来描述。这种方法产生自旋纯的双偶态,并且不存在自旋污染问题。此外,除了单激励外,还考虑了双激励,以避免在描述锥形交叉口时可能出现的问题。根据不同活动空间激发能的试验计算选择活动空间。SH轨迹以0.1 fs的核时间步长传播1ps。采用Granucci和Persico基于能量的退相干校正[63]。
动力学模拟的初始条件如下:对于方法(a),我们在恒定温度T=20 K下进行基态()分子动力学,这是Crandall等人[39]在实验中给出的上限温度。对于方法(b),我们使用了相同的方法,但动力学是在状态下建模的,并且T=500 K,这是质谱实验中常见的温度[61]。温度是由一个简单的速度-重标算法控制的,即在每个时间步长乘以速度。这里,是参考(期望)温度,是当前(瞬时)温度。初始速度从均匀分布中随机取样,并按比例缩放以产生参考温度。轨迹以0.5 fs的时间步长传播100 ps。然后,从这些基态轨迹中选择1000个快照,从50 ps后开始,每50 fs采样一次。随后的光动力学模拟只使用选定的基态几何(而不是基态速度)。初始速度(用于光动力学模拟)是随机生成的,并按比例缩放以产生所需的动能(见图1)。对于方法(c),通过采样阳离子的谐波ROOM3/CISD()基态的Wigner函数(在0 K时)来制备初始条件(几何形状和速度)。此外,我们对方法(a)的这些初始条件进行了测试,重新调整了维格纳速度以产生所需的动能。此外,利用ROOM3/CISD()方法计算了样品几何形状的垂直吸收光谱。之后,进行了SH模拟。考虑了四组初始激发能量不同的SH模拟:(i)对状态的激励,(ii)对状态的激励,(iii)对能量窗口(2.3,2.9)eV的激励,以及(iv)对状态的激励。对于情形(i) - (iii),通过比较各自跃迁的振荡强度,选择振荡强度最大的跃迁所对应的状态来确定单个轨迹的初始电子状态。四批SH模拟中的每批都包括大约1000个轨迹。
在(TD-)DFT水平上的计算以及PM7的计算都是在高斯16下完成的[64]。使用MNDO程序进行OMx (x=2或3)方法(包括SH模拟)以及AM1和PM3的计算[60]。绝热动力学模拟是用我们的内部代码与MNDO结合进行的。振动分辨光谱的计算采用高斯函数16。
摘要
1 介绍
2 方法
3.结果与讨论
4 总结与结论
参考文献
致谢
作者信息
道德声明
补充信息
相关的内容
搜索
导航
#####
我们首先利用半经验方法(OM2、OM3、AM1、PM3和PM7)和DFT在(U)B3LYP/ 6-31G *水平上对金刚烷中性和阳离子进行优化。所选几何参数(见图2)如表S1所示。中性离子的CC键长度相等,在AM1、PM3、OM2和B3LYP能级上为1.53 ?,在OM3和PM7能级上为1.54 ?。CH键长度在1.10 ? (B3LYP)和1.14 ? (OM3)之间变化。我们还注意到,AM1、PM3和B3LYP预测实际上彼此相等,而OM2、OM3和PM7显示的偏差高达0.02 ?。对于阳离子,所有的方法都预测了与中性相比的延伸率,这是由Jahn-Teller效应引起的[65]。具体来说,AM1和OM2为?, PM3和OM3为?, PM7为?, UB3LYP为?。CH键也被拉长了与中性和其他阳离子的CH键相比也是如此。总体而言,使用半经验方法获得的几何形状与当前和以前的DFT计算[30,65]以及实验结果[66,67]基本一致。
图2
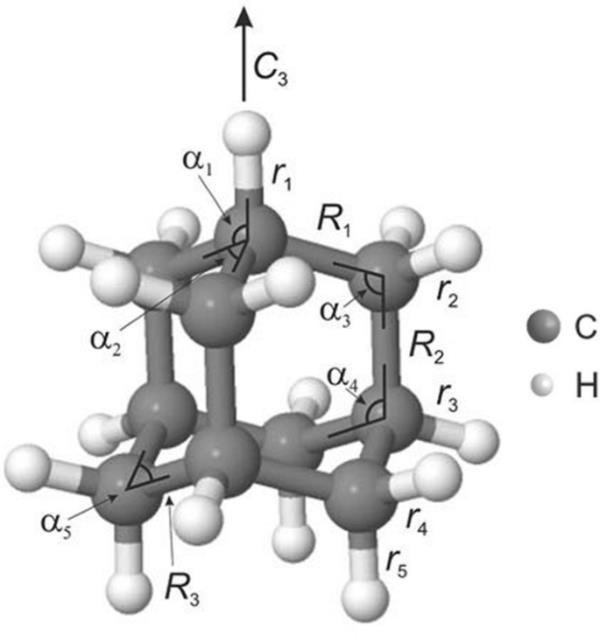
金刚烷的分子结构与标记选定的键和角度。该图改编自[30]。版权所有2018,美国物理研究所
此外,我们计算了adamantane从中性基态()到阳离子基态()的垂直(Vert IP)、绝热(Adia IP)和0-0(也称为零点能量(ZPE)校正的绝热,ZPE+)电离能(见表1)。NIST数据库中评估的实验电离能值为eV[68]。将0-0 ip与该值进行比较,我们发现PM7 (eV)性能最好,其次是OM3 (eV)。B3LYP值过小(eV)与以往的报道一致[23,30]。然而,我们选择OM3方法进行绝热()动力学模拟,因为PM7没有在MNDO软件中实现(反过来,该软件用于我们内部的绝热动力学代码),而且,OM3和PM7 0-0 ip之间的差异仅为0.08 eV。
表1 Io用半经验方法计算化势
采用TD-UB3LYP/ 6-31G *和ROOM3/CISD()方法计算了金刚烷阳离子的垂直激发能和振子强度。每种方法均适用于UB3LYP/ 6-31G *和ROOM3/CISD()优化的阳离子(最小)几何形状。激发能和振子强度收集于表2。
表2在UB3LYP/ 6-31G *和ROOM3/CISD()优化几何形状下,用ROOM3/CISD()和TD-UB3LYP/ 6-31G *计算金刚烷阳离子最低10次跃迁的激发能和振子强度
在DFT几何结构中,并简并的最低两个态和位于1.07 eV。在OM3/CISD()几何形状下,它们被发现在0.9 eV。跃迁到这些态的振子强度非常小;半经验振子强度是TD-DFT振子强度的4.3倍。接下来的两个状态,和,同样是简并的,在TD-DFT水平上位于1.8 eV,在半经验CISD水平上位于2.0 eV(这些值对两种几何形状都适用)。跃迁状态的振子强度比。同样,半经验振荡器的强度比TD-DFT的强度大3倍。从,TD-DFT与半经验CISD在状态排序上存在差异。也就是说,和在半经验水平上是简并的,而在TD-DFT水平上是简并的。然而,和之间的能隙非常小,例如TD-DFT@DFT为0.01 eV。在TD-DFT能级上,较高的跃迁(to和)位于3.8 eV,并且具有相对较大的振荡器强度。在半经验CISD能级上,跃迁是激发能低于4 eV的最后一个跃迁,且跃迁强度较大。由于有源空间的减小,和态的激发能大大提高到6 eV。然而,这些状态不会用作SH模拟的初始状态(见下文)。表S2-S4显示了主导轨道对对跃迁的贡献(用于DFT几何计算)。在这里,可以看到(i) -和状态的特征与TD-DFT和半经验CISD方法非常相似,(ii) -发生了状态重排序,(iii)和发生了明显的差异。
此外,最近Crandall等人报道了通过EPD光谱获得Ada的实验测量光谱[39]。最近,Kappe等人报道了通过光谱法在氦液滴中获得阳离子金刚烷的电子能谱[40]。这些光谱的特征是在激发能小于3.5 eV时不存在振动级数。相反,在更高的能量下,Crandall等人在宽特征的顶部观察到几个很好分辨的峰[39]。
为了计算Ada的振动分辨吸收谱,我们首先尝试使用TD-UB3LYP/ 6-31G *进行激发态优化,但失败了。这可能是因为线性响应TD-DFT势能面可能在Jahn-Teller简并附近发生翘曲[69]。在这方面,应该指出的是,本节中提供的光谱不包括非绝热效应(也见下文)。顺便提一下,我们注意到多芳烃在激发态优化中也遇到了问题,其确切原因尚不清楚[70]。因此,我们采用了一个简单的垂直梯度(VG)近似[58](它可以被视为所谓的IMDHO的一个版本,独立模位移谐振子-该模型不包括频率改变和Duschinsky旋转[71])。在TD-UB3LYP/ 6-31G *水平上计算的Ada在VG近似下的振动分辨吸收谱如图3所示。在定性上与实验一致,我们观察到两个相对较强的带,最大值分别在1.8 eV和3.8 eV,以及一个较低能量的弱带,最大值在1.1 eV。较低的能带是由向和态的跃迁构成的(它们在最小几何形状下是简并的,在VG近似下保持简并)。实验确定的能带最大值为1.12 eV[40],与计算得到的能带最大值为1.149 eV非常吻合。第一个强带是由向和态的跃迁构成的(它们也是简并的)。该带显示出大部分未解析的振动结构,除了在带的起始处有一个1.40 eV的峰值。这个峰值对应于和状态之间的0-0转换(在VG近似中)。第二个强度波段由to(简并)和to的跃迁组成。而带是无结构的,带显示出一些振动级数。与Crandall等人[39]报道的光谱在定性上一致。在定量上,该波段的第一个尖峰出现在计算光谱的3.35 eV处,而在实验光谱中它位于3.60 eV处。这个峰对应于状态和之间的0-0跃迁。
图3
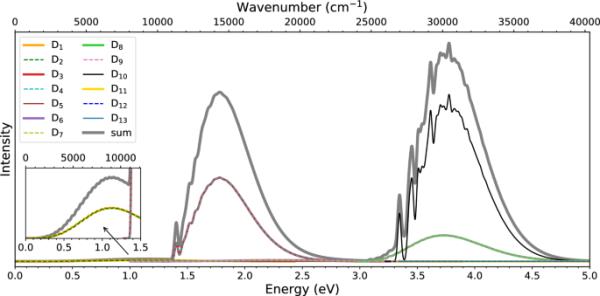
VG近似中的吸收光谱(单个贡献及其总和)。计算是在(TD-)UB3LYP/ 6-31G *理论水平上完成的。到状态,和D的转换相对较强,到其他状态的转换较弱(或非常弱)。附图显示弱带
此外,我们计算了VG近似下的光电子(光电离)谱(图4)。此外,由于我们能够使用UB3LYP/ 6-31G *优化阳离子基态(),我们计算了绝热Hessian (AH)能级的振动分辨谱[58],也称为IMDHOFAD模型[71],频率改变和Duschinsky旋转的独立模位移谐振子(见图4中的蓝色曲线)。
图4
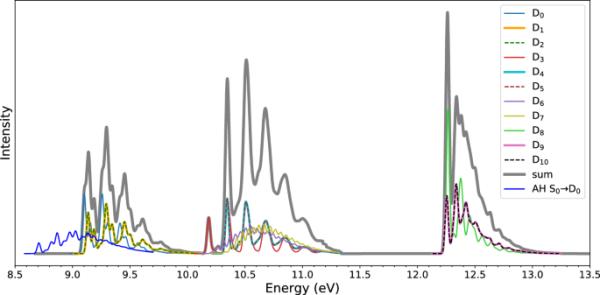
在VG近似下的光电子谱(单个贡献和它们的总和)以及在AH水平上的谱。计算是在(TD-U)B3LYP/ 6-31G *理论水平上完成的
与之前的实验[22]定性一致,我们观察到总的PE光谱中有三个波段(在14 eV以下的范围内),如图4中的灰线所示。第一个波段位于9-10 eV,包括来自,和状态的贡献。第二个波段(10-11.5 eV)包含态-。第三波段(12-13.5 eV)包括-。实验带中心分别为9.75、11.25和13.40 eV[22]。我们注意到,与上述阳离子吸收光谱相反,所有单个跃迁的PE光谱都表现出某种振动结构。
对于跃迁,我们还使用AH模型计算了频谱。与VG谱相比,AH谱是红移的,而且振动级数不同。
最后,我们从总光谱的最低波段注意到,和的0-0跃迁之间的位移仅为0.03 eV(比较图4中的“”和“/”曲线)。这个小位移已经可以从最小几何形状下阳离子的单点TD-UDFT计算中预测出来(见表S5),它给出了0.09 eV跃迁的垂直激发能。加拿大等人也报道了类似的激发能,用TD-B97X-D/ 6-311 ++G(2d,p)计算得到0.07 eV。[45]然而,我们应该注意到,最小几何形状属于对称,并且阳离子基态应该是三重简并的[72]。因此,小的光谱分裂应该是线性响应TD-DFT对地面和线性响应状态进行不平衡处理的结果[73]。
此外,我们应该强调,这里描述的光谱,除了上面讨论的近似外,并没有考虑到非绝热效应,因为金刚烷阳离子代表了jan - teller问题[29],非绝热效应对于金刚烷阳离子来说预计是相当大的。
我们模拟了能量过剩状态下阳离子的绝热动力学(见图1a,b)。具体来说,我们对方法(a)(激励度)分别以3、4和10 eV和方法(b)(激励度)分别以17、27和70 eV进行了模拟。对于后者,考虑到9-10 eV的垂直IP,给定的激发能相对于(b)分别转化为7-8、17-18和60-61 eV,提供了方法(b)的多余能量。方法(a)的3和4 eV的多余能量对应于[39]中EPD谱的较高能量部分。对于这些能量,在我们的模拟中没有观察到碎片(见下文),并且另外测试了10 eV的更高能量。对于方法(b), 70 eV对应典型EIMS实验中电子的标称动能,27 eV的能量是这些实验中实际传递给分子的能量的粗略估计(假设每个原子内部多余能为0.65 eV [61], IP为10 eV)。我们还尝试了较低的能量为17 eV,以检查方法(b)中减少能量时不会发生碎裂。
在10秒(或者更早,如果一个轨迹被终止,这发生在一些有大量过剩能量的轨迹上)的MD轨迹的最后快照被分析为碎片。计算了所有(中性和阳离子)碎片的相对丰度。对于具有唯一质量m的碎片,我们计算相对丰度为所有轨迹群中产生质量m的碎片数量与该群中产生的所有碎片总数的比率。各种过剩能量计算的相对丰度如图5所示。
图5
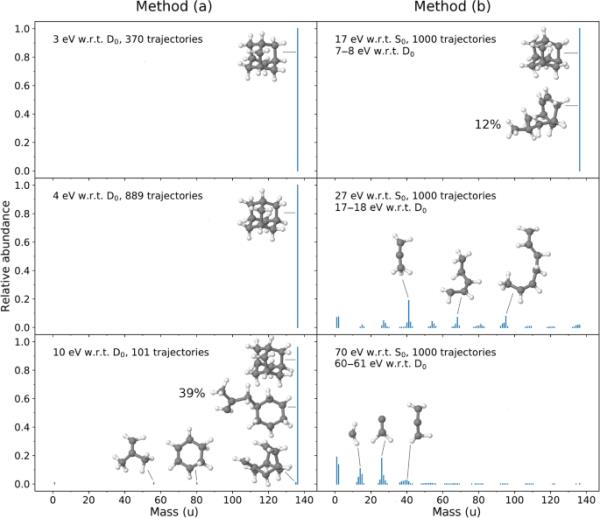
在场景(a)(左列)和场景(b)(右列)中计算的各种光子能量的相对丰度(与图1a, b相比)。所选片段也显示了出来
图5的左列对应于方法(a),右列对应于方法(b)。对于方法(a),对于3和4 eV,只观察到母离子(u)。对于能量较大的10 eV, 101个轨道中有2个出现碎片化。这两种轨迹中的一种产生了H损失,另一种显示了CH和CH的破碎(见图5,左下)。此外,39%的非碎片轨迹显示金刚烷笼的重排,如图5(左下)中六元环的衍生物所示。我们注意到,还有其他类型的重排结构,例如,如图5右上方所示的开放式笼。有趣的是,最近George和Dopfer通过红外光解光谱和量子化学计算研究了Ada的氨基衍生物金刚烷胺阳离子(Ama)的笼型开度[47,48]。研究发现,胺化在很大程度上降低了开笼反应的势垒[48],Ada的势垒从1.9 eV降至1.3 eV(包括h位移,见[47])。然而,如果有足够的多余能量,我们在模拟中观察到艾达的笼子打开。
对于方法(b), 17 eV (7-8 eV w.r.t)未观察到碎片化,但在u处有12%的母离子信号对应于重排的结构。适用于27 eV (17-18 eV w.r.t.)和70 eV (60-61 eV w.r.t),有相当多的碎片。在27 eV时,我们得到了大量的CH片段,其中n在2到10之间,而在70 eV时,较大的片段被还原成较小的片段。这里的大部分碎片在1-3个碳原子的范围内。它符合一个简单的预期,即更大的过剩能量将引起更多的碎片,碎片的尺寸将更小。
我们还应该注意到,在上述方法(a)和(b)中,我们完全忽略了Ada或Ada在激发前的动能。利用热力学的参数,我们可以从初始温度(方法(a)为20 K,方法(b)为500 K)估计出初始动能为,其中N是分子中的原子数(对于Ada或Ada)。对于K,我们得到eV,对于K,我们得到eV。新的多余能量可以通过减去旧的多余能量得到,而旧的多余能量又被定义为方法(a)和方法(b)。另一方面,通过采样Wigner函数(0 K)制备的初始条件具有动能eV。我们对方法(a)进行了修改,以使用缩放后的维格纳初始速度而不是随机生成的初始速度进行计算。考虑到初始动能约为3.1 eV, 3ev的光子能量将产生约6.1 eV的多余能量。用这些多余能量进行的模拟也显示在10ps内没有碎裂(见图S1)。
总的来说,上面描述的MD模拟(仅在状态下进行)表明,在10ps内粉碎金刚烷阳离子需要几乎10ev的多余能量。
为了模拟内部转换并明确解释阳离子激发态的影响,我们使用Tully的表面跳跃方法结合ROOM3/CISD()电子结构方法对Ada进行了非绝热动力学模拟。首先,计算了从Wigner函数中采样的几何形状的阳离子的电子垂直吸收光谱。棒谱用非归一化高斯谱展宽为:
(1)
其中,I为强度,E为激发能,分别为计算得到的激发能和振子强度,对于快照的跃迁,为展宽参数(本文为eV), M为归一化因子(此处选择M为展宽谱的最大强度)。由此计算得到的Ada的吸收光谱如图6所示。
图6
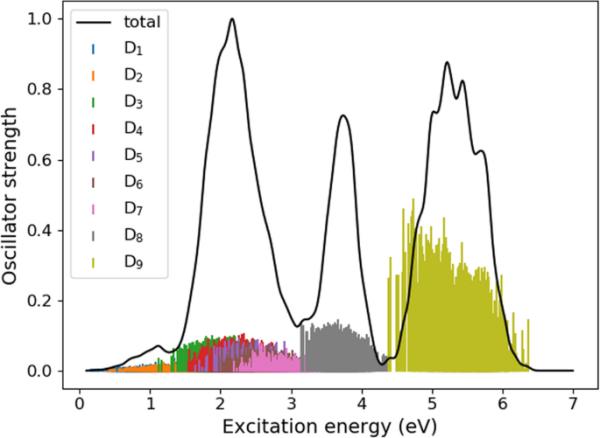
1000个轨迹初始条件的谱
在整个展宽的光谱中,我们可以识别出以下几个波段:(1)由跃迁到和组成的低能量弱波段,(2)由跃迁到-组成的强波段,(3)由跃迁产生的波段,(4)对应于的波段。我们注意到,由于初始几何形状的几何畸变,所有的跃迁都可能获得非零振子强度。与图3相比,我们可以看到,在4.5 eV以下的激发能范围内,两个光谱都呈现出两个强带。
然后,我们模拟了激发到(1)-(3)波段后的非绝热动力学。如前所述,使用ROOM3/CISD()方法时,的激发能过大。因此,我们不考虑由波段(4)激发引发的动力学。具体来说,我们考虑了四批SH模拟:(i)对状态的激励,(ii)对状态的激励,(iii)对能量窗口(2.3,2.9)eV的激励,以及(iv)对状态的激励,如方法部分所述。作为各自状态下轨迹的分数计算的电子态居群如图7所示,在前200秒内(总传播时间为1ps的居群如图7所示)。S2-S5)。我们观察到到基态()的超快内部转换。使用单一的指数函数来拟合基态种群,我们分别得到情况(i) - (iv)的时间常数为10、39、37和100 fs。因此,艾达的低洼激发态表现出极短的寿命。特别地,对于情形(iv)(激励到),使用单指数拟合,我们发现寿命fs。有趣的是,Crandall等人从他们的光谱中推断出fs的寿命[39],这是相同的数量级。我们还注意到,激发时的超短时间常数为10fs,与乙烯阳离子中电子弛豫的时间尺度(7fs)相似[74]。
图7
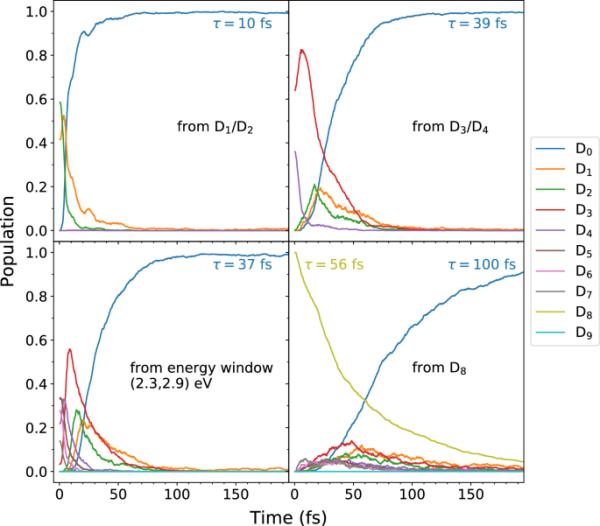
四组表面跳跃模拟的电子态居群。基态()积累的时间常数()由以下形式的单指数拟合得到;状态的寿命由这种形式的单指数拟合得到。(拟合范围为0 ~ 1ps;拟合曲线未显示。)
此外,我们分析了破碎的SH轨迹。值得注意的是,与仅在状态下的模拟相比,所有四批SH模拟都证明了在所使用的(低)激发能下超快H损失通道的可行性。然而,至少在模拟期间(1 ps), H损失产率很小,见图8。在那里,我们绘制了H损失率,定义为显示氢解离的轨迹数与轨迹总数的比值,作为激发能的函数,使用以平均激发能为中心的条,宽度为两倍标准差(初始激发能)。激发到和后,H损失率为0.9%。对于激励和,它是1.5%。同样,激发到能量窗(2.3,2.9)eV时,产率为1.8%。在激励后,我们发现H损失率为7.8%(图8)。这些数字都是指1 ps的动力学时间间隔。此外,我们发现H损失发生在基态(),即内部转换之后。此时,碎裂的次数跨越了1ps的整个间隔。图9给出了反应轨迹的示例(表示H解离)。可以看到,对于这个特定的轨迹,内部转换到基态大约需要20秒,而H损失需要额外的20秒。为了理解为什么在我们的SH模拟中H损失发生在低激发能下,我们使用ROOM3/CISD()和UOM3(含费米散射)估计了状态下的H离解能。解离能计算为CH基态最小值与拉长10 ? CH距离的结构之间的差值(如图2),在固定约束下进行优化。发现ROOM3/CISD()的解离能为1.02 eV,而UOM3(费米涂抹)的解离能为2.09 eV。CCSD(T)估计值为1.12 eV[40]。此外,我们在该状态下进行了BOMD运行,从一个反应SH轨迹的初始条件开始(启动于),并使用ROOM3/CISD()或UOM3+费米涂抹。我们在CISD水平观察到H损失,但在UOM3水平没有观察到H损失。因此,在我们的SH模拟中观察到的低能H损失可以用ROOM3/CISD()水平上较低的基态H解离能来解释。
此外,对于最大激发能(激发to),除了H损失外,我们还观察到H损失(只有2个轨迹)和CH损失(只有1个轨迹)。我们没有观察到其他更大的碎片的形成(在1ps期间)。然而,我们观察到金刚烷笼的重排。也就是说,我们发现(i)、(ii)、(iii)和(iv)批次中分别有0.3%、0.5%、1.0%和3.7%的母信号来自重排结构。最后,我们注意到,在VUV iPEPICO光谱获得的击穿图[45]中,相对于(或相对于)能量eV的母离子信号不存在。因此,相应的碎片化过程似乎比我们工作中用MD模拟解决的时间尺度要慢。
图8
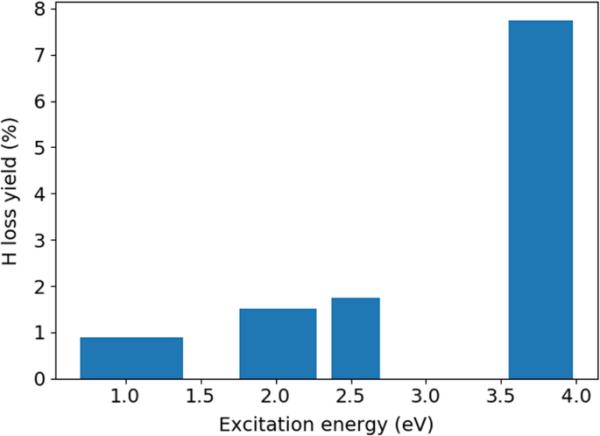
H损失产率作为激发能的函数。条形图以平均激发能为中心,宽度为标准差的两倍
图9
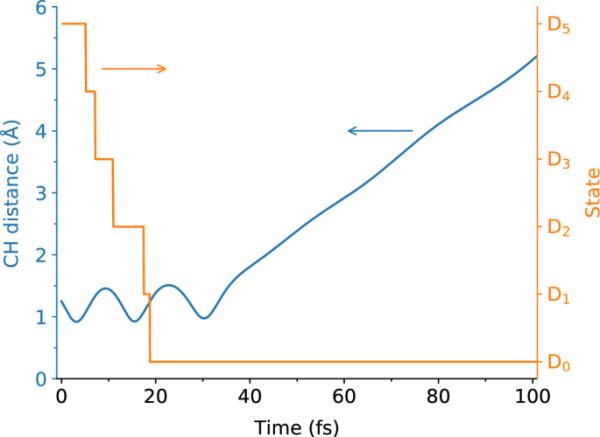
Ada的氢解离反应轨迹。所示为CH距离和当前状态作为时间的函数。轨迹取自SH批次(iii)
我们进行了金刚烷阳离子(Ada)的计算研究,解决了它的价谱和分子动力学的几个方面。首先,我们分别计算了Ada和Ada的振动分辨吸收和光电子能谱,不仅考虑了阳离子(Ada)的最低电子激发态,而且考虑了阳离子(Ada)的较高电子激发态的跃迁。这些计算是在(TD-)DFT (B3LYP/ 6-31G *)理论水平上使用垂直梯度近似完成的。计算光谱与实验光谱半定量一致。其次,我们采用半经验的OM3电子结构方法进行绝热()从头算分子动力学模拟,并提供一定的多余能量来模拟Ada在(photo)激发后的快速破碎。虽然在10 eV的多余能量中观察到相对于的快速碎片化(在低于10 ps的时间尺度上),但在较低的多余能量(在UOM3+费米散射水平上)没有检测到它。第三,应用表面跳变方法研究Ada的内部转换。采用基于OM3分子轨道的单双组态相互作用,考虑了多达10个电子态,建立了1ps的非绝热动力学模型。我们发现Ada经历了一个超快的内部转换到状态(在10-100 fs的时间尺度上,取决于初始激发能)。在一些轨迹中,内部转换之后会出现超快碎片化(主要是H损失)。具体来说,我们发现激发能为1-4 eV的H损失率为1-8%。在这种情况下,H损失的发生源于ROOM3/CISD()水平上较低的基态()H解离能。
以下是电子补充材料的链接。
下载原文档:https://link.springer.com/content/pdf/10.1007/s00214-023-03006-8.pdf











